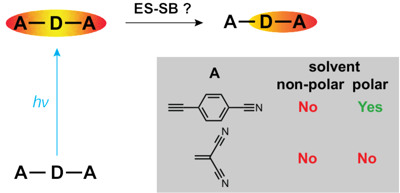
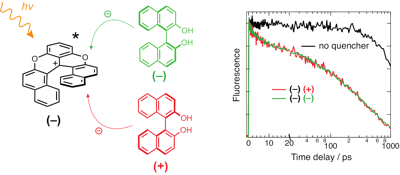
-
Photoinduced Electron Transfer between Dipolar Reactants: Solvent and Excitation Wavelength Effects
P. Verma, D.S. Budkina and E. Vauthey
Journal of Physical Chemistry B, 128 (2024), p1231-1240


DOI:10.1021/acs.jpcb.3c07922 | unige:174785 | Abstract | Article HTML | Article PDF | Supporting Info
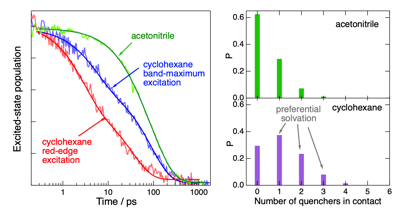
Electron transfer (ET) quenching in nonpolar media is not as well understood as in polar environments. Here, we investigate the effect of dipoleââ¬âdipole interactions between the reactants using ultrafast broadband electronic spectroscopy combined with molecular dynamics simulations. We find that the quenching of the S1 state of two polar dyes, coumarin 152a and Nile red, by the polar N,N-dimethylaniline (DMA) in cyclohexane is faster by a factor up to 3 when exciting on the red edge rather than at the maximum of their S1 ââ S0 absorption band. This originates from the inhomogeneous broadening of the band due to a distribution of the number of quencher molecules around the dyes. As a consequence, red-edge excitation photoselects dyes in a DMA-rich environment. Such broadening is not present in acetonitrile, and no excitation wavelength dependence of the ET dynamics is observed. The quenching of both dyes is markedly faster in nonpolar than polar solvents, independently of the excitation wavelength. According to molecular dynamics simulations, this is due to the preferential solvation of the dyes by DMA in cyclohexane. The opposite preferential solvation is predicted in acetonitrile. Consequently, close contact between the reactants in acetonitrile requires partial desolvation. By contrast, the recombination of the quenching product is slower in nonpolar than in polar solvents and exhibits much smaller dependence, if any, on the excitation wavelength.